Research
Stay up-to-date on the latest studies and bench-to-bedside therapies at Hopkins.
Clinical Trials
What is a Clinical Trial?
A clinical trial is a research study that involves people. Many clinical trials are performed to investigate the safety and effectiveness of new medications. However, researchers also conduct studies with people with CF to better understand basic lung biology, infections, symptoms and how all of these change over time. Download a brochure on clinical research. Trials that investigate the safety and effectiveness of medications in people are performed in a stepwise fashion. The process of testing new drugs takes several years and is separated into four phases:
Phase I clinical trials are designed to determine if a drug is safe for use in humans. These studies test a drug in a small number of people to determine the best method of giving the drug and how much can be given safely.
Phase II studies look at both safety and evidence of effectiveness. Determining the best dosing schedule is also part of a Phase II trial.
Phase III trials determine if a drug really works. As drugs progress through these three phases of clinical trials the number of patients needed increases dramatically. Phase III trials often require hundreds if not thousands of volunteers. These so-called multi-center trials are performed at several research centers at the same time.
Phase IV trials are performed immediately before as well as after a drug has been approved for use by the Food and Drug Administration (FDA). These are often large open-label studies investigating the safety of a drug. This means that all the patients in the study receive the drug; a placebo is not used. Phase IV trials are sometimes called post-marketing trials.
If you are interested in participating in a clinical trial at Johns Hopkins, please contact our Clinical Research Office at 410-955-1167 or complete the online form.
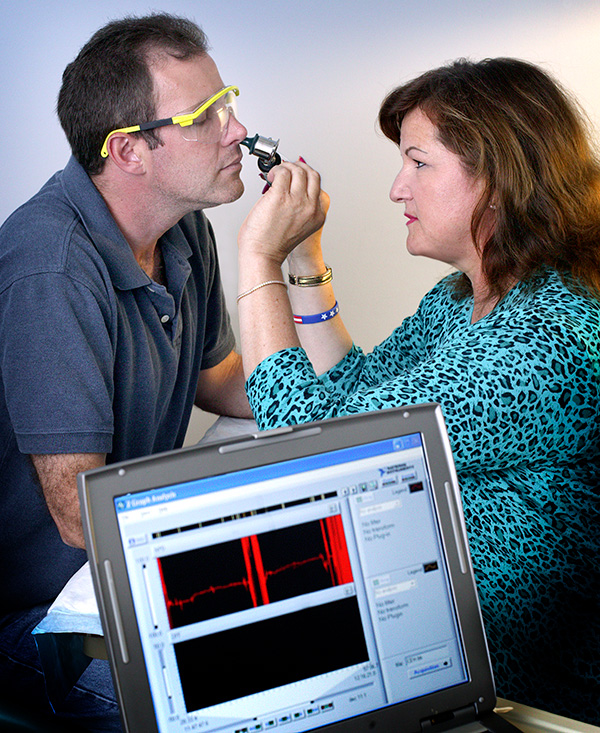
Nasal potential difference (NPD) measurement performed as part of a clinical trial.
Ongoing Trials
PI: Peter J. Mogayzel, Jr. MD, PhD, MBA
CONTACT: Mary Rykiel at 410-955-1167
PROTOCOL: IRB00243621
Contact the Research TeamPI: Peter J. Mogayzel, Jr. MD, PhD, MBA
CONTACT: Deanne Reyes at 410-955-1167
PROTOCOL: IRB00243621
Contact the Research TeamPI: Peter J. Mogayzel, Jr. MD, PhD, MBA
CONTACT: Deanne Reyes at 410-955-1167
PROTOCOL: IRB00192332
Contact the Research TeamPI: Peter J. Mogayzel, Jr. MD, PhD, MBA
CONTACT: Deanne Reyes at 410-955-1167
PROTOCOL: IRB00111304
Contact the Research TeamPI: Natalie West, MD
CONTACT: Shivani Patel at 410-502-0826
PROTOCOL: IRB00171002
Contact the Research TeamPI: Natalie West, MD
CONTACT: Shivani Patel at 410-502-0826
PROTOCOL: IRB0025740
More information – Armata Subject Flyer JHU
Contact the Research Team